david.molik.co

a personal webpage.
About Me
[ About Me ][ Curriculum Vitae ][ Contact ][  ORCID ][
ORCID ][ ![]() Github ]
Github ]
About me: I’m a Computational Biologist specializing in Arthropod Genomics currently working for the United States Department of Agriculture, Agriculture Research Service, Pacific Basin Agricultural Research Center, Tropical Pest Genetics and Molecular Biology Research Unit (USDA ARS PBARC TPGMBRU). I’m currently working on Heterochromatin Computational Biology Software, to build more accurate and useful genomics for research in combatting agricultural insect pests. Before my position with the USDA ARS, I was working on my thesis at the University of Notre Dame, before that, working as a Scientific Informatics Developer at Cold Spring Harbor Laboratory.
I currently work on Genomic aligners and polishers for non-model systems. This includes creating new cutting-edge software that can overcome the unique hurdles that non-model systems present.
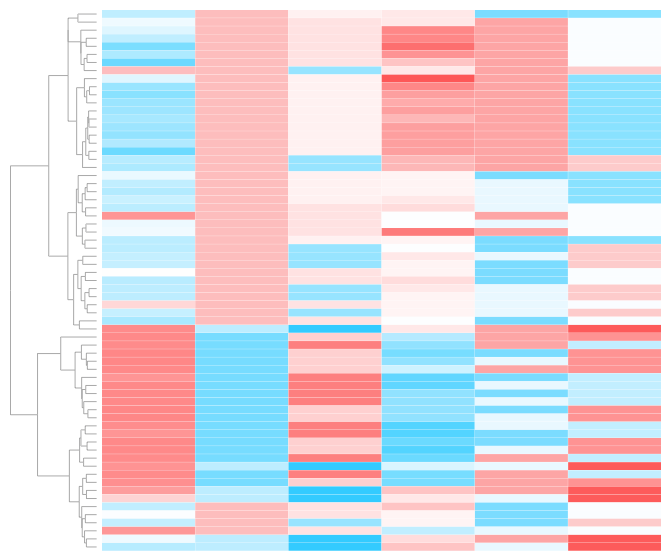
environmental variables influencing microbiome sample to sample simularity
I’ve had and continue to have a passion for teaching bioinformatics, computational biology, and software engineering to anyone who’s be interested, I started with informally teaching git at Cold Spring Harbor Labs, which evolved into designing and teaching a course ( [BIOS 60132] Introduction to Computational Genomics ) at the University of Notre Dame as a PhD, as well as designing the Introductory bootcamp for Bioinformatics at Notre Dame, Bioinformatics Introductory Program Week (BIP Week). I continue to build on my teaching skills and look for new opportunities to teach. Along with my passion for teaching, I strive to mentor others who want to learn bioinformatics, starting at Cold Spring Harbor, I’ve mentored 7 or 8 undergraduate students across a wide spectrum of backgrounds, and trained an incoming employee at Cold Spring Harbor Lab.
During my PhD, I utilized Natural Language Processing to analyze the environment that a microbe might be in, used agent-based modeling to understand the construction of metabarocdes and wrote a scientific analysis platform for analyzing microbiomes. Along with that main body of computational research, I analyzed Single Nucleotide Polymorphisms in yeast in a project that sought to understand how toxicity affected mutations. I also used data mining techniques to understand how Athropod genomes and the research around them has evolved.
My focus, and what I enjoy most in my research, is the methodology. While I care deeply about the impact that my research has on society, the thing that keeps me coming into work each day is the novelty of method I am working on, coming up with, and trying new computational analyses.
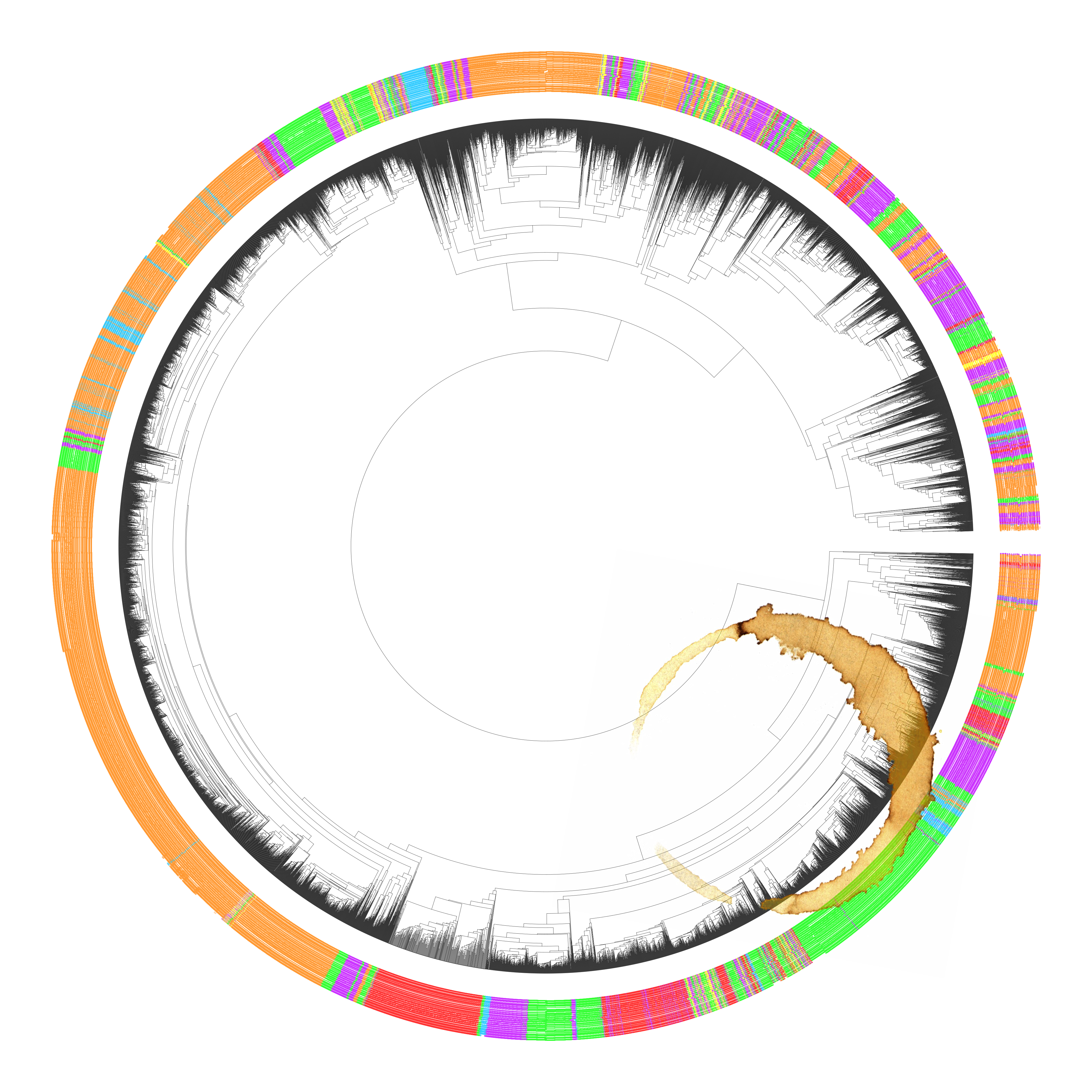
12634 18S metagenomic datasets compared to eachother, colored by type of source, ie:soil vs. aquatic, I accidentally put my coffee down on the visualization which accounts for the stain.